Cystic fibrosis:
- Cystic fibrosis (CF) is one of the genetic diseases i.e. it can be inherited to offspring.
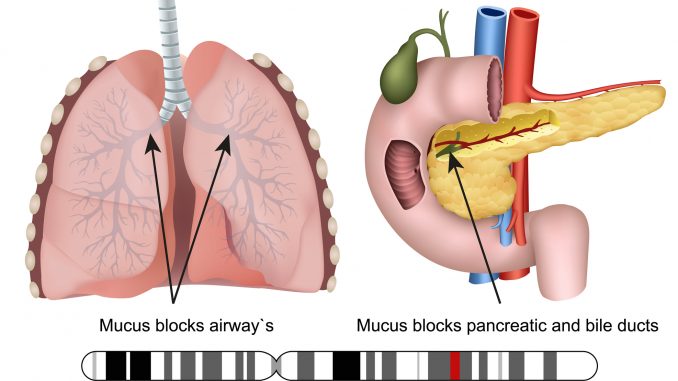
- It is the condition where the mucus produced is unusually thick and sticky that mainly affects the lungs and digestive systems along with other body organs.
- CF affects the exocrine glands such as-sweat glands, mucus secreting glands and digestive juice secreting cells.
- Normally, the mucus produced is thin and slippery but in case of CF, the defective gene leads to the creation of thick and sticky mucus.
- The so formed thick and sticky mucus blocks the respiratory ducts resulting difficult breathing and also interferes with digestive function of pancreas.
- Not only lungs and pancreas, people with CF may also face male infertility as the thick mucus causes the blockage of the Vas deferens, or epididymis preventing release of sperm from testis.
- The term Cystic fibrosis was given as the thick mucus built up inside respiratory tract that cause severe lung damage-cysts formation and fibrosis.
- CF is known to appear in almost all ethnic communities but is most common among Caucasians of Northern European descent.
genetics behind Cystic fibrosis:
- CF is genetically transferred in an autosomal recessive pattern. Each individual inherits two CFTR genes- one from each parent. Descendants who inherit a defective CFTR gene from each parent will develop CF and those who inherit one defective and one normal CFTR gene are ‘CF carriers’. Carriers usually don’t show any symptoms of CF but are capable of inheriting it to their progenies.
Causes:
- The alteration in the Cystic fibrosis transmembrane conductance regulator (CFTR) gene results Cystic Fibrosis.
- CFTR gene located on chromosome no. 7 codes for CFTR protein that functions as ion transport channels in and outside the cells related with secretion of sweat, tears, mucus, saliva and digestive juices.
- The transporter channel carries the negatively charged chloride ions which regulates the movement of water in tissues. This controlled movement of water is required for the secretion of thin and slippery mucus.
- The CFTR protein also controls other channels like transport of positive sodium ions across cell membranes that are responsible for proper functioning of lungs and pancreas.
- The deletion mutation in tenth exon of the CFTR gene known as delta f508 mutation which results in loss of phenylalanine residue from the polypeptide chain.
- This delta f508 mutation leads to the unusual production or early degradation of CFTR protein and as a result there is reduced permeability of chloride ions across CF epithelia.
- Hence, the mutation in CFTR gene alters the functioning of CFTR protein resulting Cystic fibrosis.
Symptoms:
- On the basis of severity of disorder, symptoms vary from individual to individual. Here, the classification is done as respiratory and digestive symptoms.
- Respiratory symptoms:
- Lung infections
- Stuffy nose due to inflammation of nasal passages.
- Frequent occurrence of sinusitis
- Fatigue
- Persistent coughing along with thick mucus
- Bronchitis
- Difficulty in breathing/ Wheezing
- Pneumonia
- Digestive Symptoms:
- Diarrhea or foul odor and greasy stools
- Gastritis
- Constipation
- Inflammation of pancreas
- Nausea
- Loss of appetite
- Slow growth and development in child
- Weight loss causing Malnutrition
- Bowel obstruction in new born babies.
- Other Symptoms:
- Salty test of skin due to salty sweat
- Infertility in male
- Osteoporosis
- Liver problems
- Diabetes
- Difficulty during pregnancy
Diagnosis:
- Cystic fibrosis can be diagnosed by the help of various tests:
- Immunoreactive trypsinogen test (IRT)
- IRT is a screening test performed for new born babies to detect the levels of protein called IRT in the blood.
- It is the standard test for newborn screening. The high levels of IRT denotes cystic fibrosis. However, further testing should be proceeded for confirmation of CF.
- Sweat chloride test:
- The elevated levels of chloride in the sweat is diagnosis of CF.
- Sweat chloride test is executed by using a chemical that produces sweat when triggered by relatively weak electric current.
- Sweat is accumulated either in paper or pad and then analyzed.
- Saltier sweat than normal is diagnosis for CF.
- Chest X-ray:
- Blockade of respiratory tracts causes the inflammation of lungs and this can be disclosed by chest X-rays.
- Sputum test:
- A sample of mucus is taken and it is examined to check whether it is infected or not.
- This test also shows the microorganisms present and determines the antibiotics for treatment.
- CT Scan:
- CT scan displays the detailed view of the internal structures such as pancreas and lungs.
- This helps to assess the range of damage caused by cystic fibrosis.
- Pulmonary Function tests:
- The function of lungs such as inhale and exhale of air and the transportation of oxygen to various parts is analyzed by these tests.
- Any unusual response may indicate cystic fibrosis.
- Genetic tests:
- This test is carried out by checking a sample of blood or cheek cells which checks for defective gene causing cystic fibrosis.
- This test also reveals carrier of cystic fibrosis.
Treatment:
- There is no any known cure for cystic fibrosis till date. However, treatments are prevalent for the control and management of symptoms in order to provide a quality life for individuals with CF.
- Implanted devices are used for long term administration of drugs.
- CF transmembrane conductance regulator (CFTR) modulators are latest medications to target the defective CFTR gene.
- Aerobic exercise is advised that involves harder breathing for the release of mucus from airways.
- Air passage clearance:
- It is very difficult to get rid of mucus for individuals with CF and they have hard time breathing.
- In order to clear the mucus and allow them clear breathing, airway clearance techniques (ACT) are used.
- ACT also minimizes lung infection. For instance, a therapist strokes the chest and help to free mucus.
- Also oral medications are prescribed to thin the mucus, mobilize it and kill germs.
- Azithromycin and ibuprofen are prescribed for enhancing lung function.
- Bronchodilators help to clear the airways by relaxing muscles around them.
- Nutritional therapy:
- CF is responsible for the disturbance in digestive system and absorption of nutrients.
- Individuals with CF need to discuss about proper diet with the dietician.
- Pancreatic or other digestive supplements if needed should be balanced for proper digestive absorption.
- High calorie and high fat diet is recommended for children with CF for their proper growth and development.
References:
- https://www.medicalnewstoday.com/articles/147960#treatment
- https://www.mayoclinic.org/diseases-conditions/cystic-fibrosis/symptoms-causes/syc-20353700
- https://www.webmd.com/children/what-is-cystic-fibrosis#1
- https://www.medicinenet.com/cystic_fibrosis/article.htm#how_is_cystic_fibrosis_treated
- https://www.healthline.com/health/cystic-fibrosis#treatments
- https://ghr.nlm.nih.gov/condition/cystic-fibrosis#diagnosis
- https://www.nhs.uk/conditions/cystic-fibrosis/